Evaluate prior specifications of a joint BLRM for oncology dose finding
intro_jointBLRM.RmdThis vignette illustrates the usage of the decider
package for setting up and evaluating prior distributions for joint
Bayesian logistic regression model (BLRM)-based dose-finding trials with
one or more monotherapy and/or two-compund combination therapy trials
that may run sequentially or in parallel. It will be assumed that the
reader is familiar with the general process of dose-limiting toxicity
(DLT)-based dose-finding trials with the aim of determining a
maximum-tolerated dose (MTD) and with the BLRM and its meta-analytic
extensions (here referred to as joint BLRM) for DLT-based dose finding.
For an introduction to these topics, refer to (Neuenschwander, Branson, and Gsponer 2008),
(Neuenschwander et al. 2014), (Neuenschwander, Roychoudhury, and Schmidli
2016). While this terminology and the applied methods mainly
originated in the context of oncology dose finding, the methods
contained in this vignette can also be applied to other therapeutic
areas. Note that different therapeutic areas may be using a slightly
different notions instead of e.g. DLT and MTD.
Note further that the term “trial” may also refer to a trial arm (e.g. monotherapy and combination therapy), and that the model will not differentiate between these notions. Moreover, the term “study” will refer to the complete conduct of all jointly modeled trials. That is, simulating e.g. 10 studies means that all involved trials are simulated in the specified order and under the remaining specified assumptions for in total 10 times.
Overview of the main tools
The package ships with two main functions, which are called
scenario_jointBLRM and sim_jointBLRM. The
former is essentially a high-level wrapper for fitting a joint BLRM to a
concrete data scenario, while the latter allows to simulate the course
of a number of dose-finding trials under the assumption of a specific
dose-toxicity scenario, i.e. a specification of the DLT rates of the
involved dose levels.
scenario_jointBLRM
The function scenario_jointBLRM is essentially a wrapper
for a Stan model of the joint BLRM, which is internally used to sample
from the posterior distribution given a set of input data using the
so-called No-U-Turn sampler (NUTS) algorithm. For details on the
technicalities behind Stan, a good starting place is Stan’s homepage.
However, besides fitting the model, the function will also perform a number of further evaluations based on the posterior. Most importantly, the posterior distribution of the DLT rates (referred to as posterior toxicities in the following) for a given set of dose levels is determined based on the fitted model. In actual dose-finding trials these distributions will form the basis of the recommended dose level for the next cohort.
The output of scenario_jointBLRM consist of a summary of
these posterior toxicities, as well as an evaluation of them in terms of
an specific escalation rule. Here, the term escalation rule refers to a
method that derives a dosing recommendation based on the fitted BLRM.
Currently, the decider package supports the popular
escalation with overdose control (EWOC)-based escalation rules (Babb, Rogatko, and Zacks 1998), as well as
loss-based escalation rules, which are however not considered in this
vignette. Note that EWOC-based escalation is the default setting of all
functions, so that without further specifications, the functions will
assume that EWOC is applied. Essentially, EWOC computes the probability
that the DLT rate of a dose lies above a prespecified target dosing
interval (often from 16% to 33% DLT rate), and demands that this
probability does not exceed a certain boundary, typically 0.25 (the
default). Both the interval and the boundary can be adapted by the user.
For EWOC-based escalation, the summary that
scenario_jointBLRM creates will contain the resulting
posterior interval probabilities of underdosing, target dosing, and
overdosing, as well as summary statistics of the posterior of the DLT
rates of a given set of doses of interest (which are usually the set of
possible dose levels in the trial).
Additionally, plots of the posterior interval probabilities can be created, which visualize the EWOC principle and the set of doses that is allowed by EWOC based on the given data.
The main uses of scenario_jointBLRM are the evaluation
of actual and hypothetical data scenarios and the determination of the
resulting posterior. Actual data scenarios refer here to data that was
observed during a trial, while hypothetical data scenarios are a way of
evaluating the on-trial behavior of the BLRM with some escalation rule
in a concrete setting. These are particularly important at the planning
stage of dose-finding trials, as they allow to monitor the performance
of the model in scenarios that are assumed to potentially occur during
the trial. Based on these evaluations, the prior distribution of the
BLRM can be adapted to obtain appropriate escalation behavior.
For evaluating a prior specification at the planning stage of a
trial, one will typically need to consider a whole set of different
scenarios that test the model and its recommendation in a broad spectrum
of possible situations. Specifically for this setting, the additional
function scenario_list_jointBLRM() is provided, which
allows to evaluate a list of arbitrarily many scenarios via
scenario_jointBLRM() in parallel.
sim_jointBLRM
When a prior with appropriate on-trial behavior is established, one may want to conduct more thorough evaluation of the performance of the resulting model. A widely-accepted way of achieving this are trial simulations. For this, one assumes concrete “true” DLT rates for the dose levels available in a trial (referred to as dose-toxicity scenario) and repeatedly simulates the number of DLTs that are observed in cohorts of patients. Based on this data, the BLRM is fitted and the escalation rule is applied to determine the dose level for the next cohort, or, potentially, if the trial needs to be stopped early (this can e.g. happen when the lowest available dose does not satisfy the EWOC criterion). This process is repeated until a prespecified condition for the declaration of the MTD is reached, or until the trial is stopped without declaring an MTD (either due to stopping early or due to reaching the maximum number of patients that can be enrolled).
Such trial simulation can then be used to determine the operating characteristics of the trial design in specific dose-toxicity scenarios. That is, one simulates a number of trials, e.g. 1000, and determines the ratio of MTDs declared at specific dose levels (e.g., MTDs at target doses), as well as further quantities like the average number of patients treated with a specific dose or the average number of DLTs that occurred during the trial.
This process is then repeated for a number of different scenarios, containing e.g. low-toxicity scenarios (all doses have DLT rate below the targeted interval), moderate scenarios (DLT rates range from underdoses over target doses to overdoses), highly toxic scenarios, and further extreme cases, like e.g. one trial being considerably more toxic than another one.
The function sim_jointBLRM() is designed to perform
these simulations in a convenient manner and generate output tables that
summarize the operating characteristics of a specified trial design in
concrete dose-toxicity scenarios. It provides the option two simulate up
to six trials running in parallel (which can be activated independently
of each other), namely two monotherapy trials for each involved
compound, and two combination therapy trias which are all included in a
joint meta-analytic BLRM which is specified according to (Neuenschwander et al. 2014), (Neuenschwander, Roychoudhury, and Schmidli
2016).
The priors used by the model and the order in which cohorts arrive can be freely specified by the user, as well as many further options, including escalation rules, MTD selection, starting doses, and additional restrictions on the increment between escalations. Furthermore, the function allows to include data from arbitrarily many historical trials (not actively simulated) for the involved compounds.
Setting up and evaluating priors using
scenario_jointBLRM
Assume in the following that we are interested in setting up an oncology dose-finding study that includes two different trials, referred to as Arm A and Arm B in the following. Suppose that Arm A investigates on some treatment, say compound 1, in a first-in-human monotherapy setting, while Arm B shall consider combination therapy with compound 1 and some combination partner, say compound 2. We assume that previous dose-finding trials for compound 2 were already conducted, so that DLT data is available for monotherapy with compound 2 from earlier clinical investigations.
Assume further that Arm A and Arm B shall run partially in parallel,
or, more precisely, that Arm B will be initiated before Arm A has
concluded (before the trial declared an MTD), provided that the involved
doses of compound 1 were already tested in monotherapy. To allow
exchange of information across Arms A and B, and further the inclusion
of the historical data for compound 2, we will model both trials and the
historical trials in a joint BLRM. The following sections provide an
overview how the function scenario_jointBLRM can be set up
for this situation, and how it is used to specify and evaluate an
appropriate prior distribution and conduct analyses to determine the
next dose level during the trial.
Before you continue, do not forget to load the decider
package.
Basic specifications of scenario_jointBLRM
As a first step, we will determine the necessary fixed input data, consisting of a set of available dose levels for Arms A ad B, the set of historical data, and reference dose levels for both compounds.
First, assume that from an historical monotherapy trial for compound 2, the clinical data given below is available. Dose levels of the same compound are assumed to be stated in some fixed unit of measurement, e.g. mg or mg/kg, which is not explicitly denoted for simplicity.
| Dose | #Patients | #DLT |
|---|---|---|
| 2 | 3 | 0 |
| 4 | 3 | 0 |
| 8 | 3 | 0 |
| 12 | 9 | 1 |
| 16 | 12 | 2 |
It is further assumed that all of these observations originate from a single historical trial, although this could easily be extended to multiple historical trials (described below).
The given data now needs to be written in the format that
scenario_jointBLRM accepts, which is a named list
containing the entries dose1, dose2,
n.pat, n.dlt, and trial. For the
data given above, use
historical.data = list(
dose1 = c( 0, 0, 0, 0, 0),
dose2 = c( 2, 4, 8, 12, 16),
n.pat = c( 3, 3, 3, 9, 12),
n.dlt = c( 0, 0, 0, 1, 2),
trial = c("H1", "H1", "H1", "H1", "H1")
)Here, each column in the structure given above translates to the data
observed at one dose level during a specific historical trial. Note that
the dose levels of compound 2 are given in the entry dose2,
while the entry of dose1 is 0 to indicate that
the cohorts received monotherapy with compound 2.
In the example here, only the data from one historical trial is
included. For this, the same entry of historical.data$trial
is given for each historical cohort, which tells the function that all
of these cohorts belong to the same trial. The entry "H1"
is an arbitrarily chosen trial name for the historical trial, note that
one could also use a specific number or some other string as trial name.
When data for multiple historical trials in given, multiple trial names
need to be used to indicate this.
As a next step, we need to specify reference dose levels and the set of available dose levels for the trial. Although the set of investigational dose levels can be flexible in practice (i.e., does not need full specification at the planning stage), for prior evaluations, a hypothetical set of doses that are planned to be used in the trial need to be specified to allow evaluating the hypothetical on-trial behavior of the model.
We will in the following assume that the planned monotherapy doses for compound 1 are
d1 = c(0.1, 0.2, 0.4, 0.8, 1.6, 2.4, 3.6, 5, 6)where we have again left out the unit of measurement (e.g. g or mg/kg) for simplicity. Regarding combination therapy, suppose that it is planned to consider all combinations of the monotherapy doses of compound 1 with the dose levels
d2 = c(8, 12)of compound 2 (given in the same unit of measurement as the doses in the historical data).
The planned monotherapy and combination therapy doses should be given
to the function via the argument doses.of.interest, which
specifies which dose levels need to be evaluated. The argument needs to
be a matrix with two rows and one column for each dose level, where
monotherapy doses are indicated by placing 0 or
NA in the entry of the other compound.
In the setting specified above, this results in
doses.of.interest = rbind( c(d1, rep(d1, times=length(d2))),
c(rep(0, length(d1)), rep(d2, each=length(d1))) )
#monotherapy compound 1.
doses.of.interest[, 1:length(d1)]
#> [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9]
#> [1,] 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6
#> [2,] 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0 0
#combinations of compound 1 with dose 8 of compound 2.
doses.of.interest[, (length(d1)+1):(2*length(d1))]
#> [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9]
#> [1,] 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6
#> [2,] 8.0 8.0 8.0 8.0 8.0 8.0 8.0 8 8
#combinations of compound 1 with dose 12 of compound 2.
doses.of.interest[, (2*length(d1)+1):(3*length(d1))]
#> [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9]
#> [1,] 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6
#> [2,] 12.0 12.0 12.0 12.0 12.0 12.0 12.0 12 12Next, we need to specify the reference doses used by the joint BLRM.
We assume that for compound 1 the dose 6 and for compound 2
the dose 12 was selected for this:
dose.ref1 = 6
dose.ref2 = 12To explicitly tell the function that we are interested in two trials which consider monotherapy for compound 1, respectively combination therapy, we now include the following optional arguments:
The trial names "A" and "B" are arbitrary
and tell the function that this is how we will indicate the two trials
in the data. The types "mono1" and "combi"
specify that only the doses.of.interest for the
corresponding trial type need to be analyzed for the corresponding
trial. That is, trial "A" is the trial of type
"mono1" and will only consider monotherapy with compound 1,
while "combi" indicates that trial "B" will
only consider combination therapy doses. There is also the type
"all" which will cause the function to analyze all given
doses of interest for a trial.
If the trials of interest or their type are not explicitly specified,
the function will use all trial names given in data and historical data
(in our case, including the trial "H1" from the historical
data), and further try to infer the type from the given data for this
trial. If this is not possible, (e.g. when no data is given yet), the
function will provide posteriors for all doses of interest for this
trial. Therefore, specifying trials of interest and their types allows
to control which output is included in a more precise fashion.
Furthermore, when we do not provide data for trial "A"
or "B" yet (e.g. to obtain the predictive distribution
induced by the data from the other included trials), the function would
not include the posterior of the trial in the output unless its name is
specified explicitly as a trial of interest. For instance, when only the
historical data is given, the function would only estimate the posterior
of the historical trials but not estimate the predictive distribution of
a new trial based on the historical data.
At this point, we could in principle already run the function to
produce posterior predictive distributions for the trials
"A" and "B" purely from the historical data of
trial "H1" (which is equivalent to a meta-analytic
predictive prior, cf. (Neuenschwander,
Roychoudhury, and Schmidli 2016)). However, we still have not
specified a (hyper-)prior distribution for the model, so that the
function would apply the default prior configuration (refer to the
documentation of scenario_jointBLRM). While the default
prior is weakly informative and was found to be quite flexible across
different situations, it is still recommended to specify a prior
manually to ensure good on-trial behavior of the model This will be the
subject of the next section.
Specifying a prior for the joint BLRM
Refer to the section Detail in the documentation of
scenario_jointBLRM for a model specification of the joint
BLRM and explanations what the model does. For the following, it is only
important to know that the prior specifications consist of two parts,
namely, a prior for the hyper-means (argument prior.mu) and
a prior for the between-trial heterogeneities (argument
prior.tau), where the hyper-mean is the shared mean of the
parameters across trials and the between-trial heterogeneity is the
standard deviation across trials. Recall the five different parameters
of the joint BLRM: two parameters for each compound (more precisely, the
intercept \(log(\alpha_i)\) and the
log-slope \(log(\beta_i)\) for compound
\(i\)), and the interaction parameter,
\(\eta\). Hence, the prior
specification must specify the distribution of the hypermean and
between-trial heterogeneity for these five parameters.
For this, it is assumed that all hypermeans follow normal distributions with some fixed mean and SD (which are therefore the input required to specify the distribution of each hypermean), while the heterogeneities are assumed to follow log-normal distributions, whose mean and SD (on log-scale) need to be given to the function. Technically, it is further assumed that for each \(i\), \(log(\alpha_i)\) and \(log(\beta_i)\) may be correlated with some correlation coefficient \(\rho\), but this hyperparameter is automatically assigned a uniform distribution on the interval \([-1, 1]\), so that its prior does not need to be specified.
Prior for the hypermeans: prior.mu
First, consider the prior for the hypermeans, which is encoded in the
argument prior.mu. This must be a list with five named
entries, where each entry provides the mean and SD of the distribution
of one of the five hypermeans.
The expected format of this argument is:
# Parameter Mean SD
prior.mu = list(mu_a1 = c(-0.7081851, 2),
mu_b1 = c(0, 1),
mu_a2 = c(-0.7081851, 2),
mu_b2 = c(0, 1),
mu_eta = c(0, 1.121))Here, the prefix mu indicates that this is a hypermean,
while the suffixes correspond to the parameters as given in the
following table.
| Parameter | Suffix |
|---|---|
| \(log(\alpha_1)\) | _a1 |
| \(log(\beta_1)\) | _b1 |
| \(log(\alpha_2)\) | _a2 |
| \(log(\beta_2)\) | _b2 |
| \(\eta\) | _eta |
In the prior list, each of the entries should be of the form
c(x, y), where x gives the mean and
y the SD of the normal distribution assumed for the
hypermean of the corresponding parameter.
The default values were oriented towards (Neuenschwander et al. 2014) and (Neuenschwander, Roychoudhury, and Schmidli 2016), and represent a relatively flexible, weakly informative prior distribution. It is recommended that this is used as a starting point for adapting the prior to a given setting, as specific modifations to match the setting of a concrete trial will most likely provide better performance than the default prior.
Regarding the interpretation of the parameter, \(log(\alpha_i)\) gives the median log-odds
of experiencing a DLT following treatment at the reference dose. The
value -0.7081851 corresponds to
logit(0.33)
#> [1] -0.7081851which means that one assumes the reference dose to have in median a DLT rate of 33%. This corresponds precisely to the upper boundary of the default target interval (from 16% to 33%), so that the prior expresses the assumption that the reference dose is a priori a possible candidate for the MTD (note that the reference dose should be chosen accordingly).
The default value of the SD of the hypermeans of \(log(\alpha_i)\) is 2, which
represents a relatively large uncertainty and leads to relatively
flexible prior. Note however that depending on the dosing range and
other specifications of the trial, one might want to deviate from this
default value. For instance, we may want to assume a slightly larger
uncertainty regarding the intercept to obtain a more conservative setup,
e.g. via
prior.mu = list(mu_a1 = c(-0.7081851, 3),
mu_b1 = c(0, 1),
mu_a2 = c(-0.7081851, 3),
mu_b2 = c(0, 1),
mu_eta = c(0, 1.121))Regarding the hyperpriors for \(log(\beta)\), a standard normal
distribution is assumed. This covers a relatively broad range of slopes,
although one may want to select a larger or smaller SD in certain cases.
Regarding the mean, it is typically not necessary to deviate from the
default of 0 unless there is very concrete information
about the shape of the dose-toxicity relationship.
For the interaction parameter, recall that negative values of \(\eta\) express a lower DLT rate of dose combinations than the interaction free-case (in which the compounds would cause DLT fully independently from each other), while positive values lead to a larger DLT rate compared to the interaction-free case. The default value of \(0\) for the mean expresses that both positive and negative interactions may occur, while the SD of 1.121 is selected so that the 95% credible interval of the interaction-parameters corresponds to an increase up to 9-fold (or decrease by factor of 9) of the odds for a DLT at the reference dose. Larger or lower values of mean and SD can be used to obtain more aggressive or conservative behavior of combination trials. Refer e.g. to (Neuenschwander et al. 2014) for more detail on this.
Prior for the between-trial heterogeneities:
prior.tau
The prior parameters for the between-trial heterogeneity control the amount of borrowing that is performed across trials. In this context, the assumption of a larger heterogeneity for a parameter allows this parameter to show larger variance across trials, and, consequently, reduces the influence that data from other trials have on each jointly modeled trial.
The decider package assumes that the between-trial heterogeneities follow log-normal distributions, which are specified by providing their mean and SD on log-scale.
The default value and required format of prior.tau
is:
# Parameter Mean SD
prior.tau = list(tau_a1 = c(log(0.25), log(2)/1.96),
tau_b1 = c(log(0.125), log(2)/1.96),
tau_a2 = c(log(0.25), log(2)/1.96),
tau_b2 = c(log(0.125), log(2)/1.96),
tau_eta = c(log(0.125), log(2)/1.96))The naming conventions are the same as for prior.mu.
Note that a value of c(x,y) as tau_[...]
provides the mean x and SD y on log-scale,
i.e. if the corresponding hyperparameter is denoted as \(\tau\), the function assumes that \(\tau=exp(\psi)\) for some random variable
\(\psi\) that is normally distributed
with mean x and SD y.
The default values are oriented towards (Neuenschwander et al. 2014). Note that the SDs
are typically all set to either log(2)/1.96 or
log(4)/1.96 (larger uncertainty), other values are usually
not needed.
To specify the mean for the between-trial heterogeneities, one
typically assumes a specific degree of heterogeneity (cf. table below).
By default, scenario_jointBLRM assumes the mean to be
log(0.25) for the between-trial heterogeneity of \(log(\alpha_i)\), and
log(0.125) for the heterogeneity of the remaining
parameters. On natural (non-log) scale, this means that differences of
the parameters across trials are in median expected to be 0.25,
respectively 0.125. This is usually referred to as “moderate”
between-trial heterogeneity, other potential values for the mean can be
found in the following table.
| Heterogeneity | \(log(\alpha_i)\) | \(log(\beta_i)\) / \(\eta\) |
|---|---|---|
| Small | log(0.125) |
log(0.0625) |
| Moderate | log(0.25) |
log(0.125) |
| Substantial | log(0.5) |
log(0.25) |
| Large | log(1) |
log(0.5) |
Regarding the prior setup for the between-trial heterogeneity, the chosen prior should depend on the expected differences across trials and the number of patients in the included co-data (larger number of patients may require more heterogeneity to ensure sufficient robustness). For many situations, the moderate and substantial configurations from the above table provide acceptable robustness, so that these are typically considered to be good starting points for prior evaluations. Besides the evaluations of hypothetical scenarios and simulations (covered in the following sections), prior effective sample sizes also provide a good way of evaluating the influence of co-data on the trial of interest and adjusting the priors for the between-trial heterogeneity accordingly. Refer to (Neuenschwander et al. 2020) for more detail on this type of method for assessing the information included in the prior.
Using scenario_jointBLRM to evaluate hypothetical data
scenarios
Hypothetical data scenarios evaluate the decisions of the BLRM in a number of concrete situations that might occur in the trial that is planned. While one can usually not foresee and evaluate all possible situations (mostly due to there being too many of them), one restricts these evaluations to important classes of settings. These include but are not limited to:
Early scenarios that assess the decisions within the first few cohorts of a trial, especially scenarios that assume early DLTs.
Late scenarios that assess the resulting MTD estimates based on the planned number of patients (or slightly fewer).
Scenarios that assess specific critical dosing recommendations, e.g. whether the escalation rule ceases to increase the dose when observing certain DLT rates.
Scenarios that assume heterogeneous co-data. Here, it is e.g. assessed whether the decision rule can correctly decide in settings where the involved trials or trials show substantially different toxicities.
Similarly to the previous point, if multiple parallel trials are involved that start at different points in time, it should be assessed how the co-data influences the start of the trials. For instance, it can happen that the combination trial is not allowed to be started at the planned starting dose due to toxic monotherapy data. It should therefore be assessed (and aligned with ones recommendations) which co-data is needed to allow the planned starting dose, and in which cases this would not be recommended by the model.
As an example, consider the scenario that the first cohort of 3 patients was treated at the dose 0.1 of compound 1 and a second cohort at the dose 0.2. We assume further that no DLT occurred in the first cohort, while one of the patients of the second cohort experienced DLT. This translates to the data scenario:
scenario1 = list(
dose1 = c( 0.1, 0.2),
dose2 = c( 0, 0),
n.pat = c( 3, 3),
n.dlt = c( 0, 1),
trial = c("A", "A")
)Note that "A" was used as trial name for the monotherapy
trial for compound 1, as this needs to match the name specified in the
trials.of.interest argument.
Now, the scenario function can be called. Using the previously specified objects, the function can be applied via
result1 <- scenario_jointBLRM(
data = scenario1,
historical.data = historical.data,
doses.of.interest = doses.of.interest,
dose.ref1 = dose.ref1,
dose.ref2 = dose.ref2,
trials.of.interest = trials.of.interest,
types.of.interest = types.of.interest,
prior.mu = prior.mu,
prior.tau = prior.tau
)
print(result1)
#> $`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.11859 0.11397 0.00277 0.08298 0.42437 0.73012 0.20692 0.06296
#> 0.2+0 0.16858 0.14123 0.00622 0.13118 0.52599 0.57770 0.28426 0.13804
#> 0.4+0 0.23990 0.18077 0.01177 0.19979 0.66828 0.41439 0.30802 0.27759
#> 0.8+0 0.32845 0.22607 0.01943 0.28842 0.82258 0.28763 0.27302 0.43935
#> 1.6+0 0.42193 0.26338 0.02855 0.39217 0.92793 0.19970 0.22465 0.57565
#> 2.4+0 0.47413 0.27850 0.03468 0.45659 0.96162 0.16412 0.19714 0.63874
#> 3.6+0 0.52269 0.28863 0.04127 0.52290 0.98040 0.13637 0.17219 0.69144
#> 5+0 0.55892 0.29362 0.04641 0.57564 0.98885 0.11860 0.15342 0.72798
#> 6+0 0.57778 0.29531 0.04980 0.60451 0.99195 0.11001 0.14404 0.74595
#>
#> $`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.18629 0.13531 0.02394 0.15155 0.53989 0.52862 0.33361 0.13777
#> 0.2+8 0.23094 0.15907 0.03199 0.19187 0.63372 0.40750 0.36679 0.22571
#> 0.4+8 0.29299 0.18960 0.04220 0.25081 0.74998 0.28991 0.35425 0.35584
#> 0.8+8 0.37122 0.22352 0.05499 0.33055 0.86179 0.19460 0.30452 0.50088
#> 1.6+8 0.45729 0.25390 0.06750 0.43033 0.94195 0.13229 0.24075 0.62696
#> 2.4+8 0.50649 0.26808 0.07326 0.49341 0.96861 0.11074 0.20394 0.68532
#> 3.6+8 0.55217 0.27991 0.07487 0.55814 0.98426 0.09771 0.17248 0.72981
#> 5+8 0.58535 0.28891 0.07207 0.61212 0.99170 0.09375 0.15140 0.75485
#> 6+8 0.60188 0.29425 0.06797 0.64102 0.99437 0.09407 0.14083 0.76510
#> 0.1+12 0.22163 0.13567 0.04560 0.19111 0.56602 0.39149 0.42356 0.18495
#> 0.2+12 0.26436 0.15705 0.05503 0.23029 0.65521 0.29229 0.42656 0.28115
#> 0.4+12 0.32378 0.18514 0.06661 0.28640 0.76281 0.20083 0.38479 0.41438
#> 0.8+12 0.39874 0.21714 0.07997 0.36279 0.86979 0.13339 0.31136 0.55525
#> 1.6+12 0.48109 0.24729 0.09018 0.45862 0.94615 0.09510 0.23439 0.67051
#> 2.4+12 0.52780 0.26299 0.09012 0.52017 0.97151 0.08508 0.19534 0.71958
#> 3.6+12 0.57027 0.27851 0.08220 0.58428 0.98658 0.08529 0.16244 0.75227
#> 5+12 0.59971 0.29270 0.06724 0.63718 0.99349 0.09417 0.14046 0.76537
#> 6+12 0.61346 0.30184 0.05647 0.66692 0.99585 0.10270 0.12933 0.76797Note that the output contains two entries, one for each trial of
interest. As we previously specified the types of these trials, the
posterior toxicities of trial "A" are only computed for the
doses of interest that represent monotherapy with compound 1, while the
posteriors for trial "B" only contain the combination
therapy dose levels.
For each of these doses, the interval probabilities are computed, as
well as summary statistics of the posterior toxicities. By default, the
considered interval probabilities assume a target interval (range of DLT
rates considered to be appropriate as MTD) of \([0.16, 0.33)\). However, if one is
interested in some different interval, say, \([0.2, 0.3)\), this can be achieved by
supplying the optional argument
dosing.intervals = c(0.2, 0.3). In this context, the
underdosing, respectively overdosing intervals contain the DLT rates
below, respectively above the target interval.
Further customization of the output can be achieved by supplying the
argument probs, which specifies the quantiles listed in the
output tables. By default, the reported quantiles are 2.5%, 50%, and
97.5%. If additional quantiles shall be computed, say, 25% and 75%, this
can be achieved e.g. with
probs = c(0.025, 0.25, 0.5, 0.75, 0.975).
If additional output plots shall be created (not required for prior
evaluation, but potentially helpful for actual analyses), one can
activate this via plot.decisions = TRUE. Usually, simple
bar plots are created by this argument for monotherapy, and a heatmap
for combination therapy. By additionally setting
plot.return = TRUE, the plots will be contained in the
results list in the form of ggplot objects. Refer to the
documentation of the ggplot2 package for detail on this
type of object. The ggplot2 package also provides a broad
range of functions that can be used to customize the returned plots
further. If plot.return is not specified, the function will
only print the plots and potentially save them if the user specified a
path for outputs. Additionally, one may want to control the number of
digits the output is rounded to, especially when considering
heatmap-type plots for combination therapy, using
e.g. digits = 3.
result1a <- scenario_jointBLRM(
data = scenario1,
historical.data = historical.data,
doses.of.interest = doses.of.interest,
dose.ref1 = dose.ref1,
dose.ref2 = dose.ref2,
trials.of.interest = trials.of.interest,
types.of.interest = types.of.interest,
prior.mu = prior.mu,
prior.tau = prior.tau,
plot.decisions = TRUE,
plot.return = TRUE,
ewoc.threshold = 0.25,
digits = 3
)The generated plots are shown in the plot panel in RStudio. To
explicitly display the returned ggplot objects, one can
access them as follows.
plot(result1a$plots$"trial-A")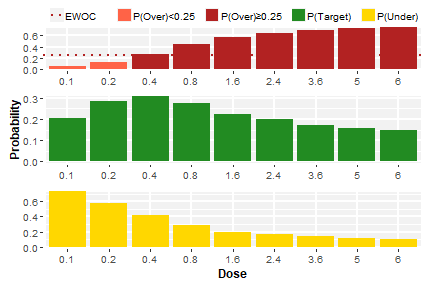
Plot generated by scenario_jointBLRM()
for trial "A"
plot(result1a$plots$"trial-B")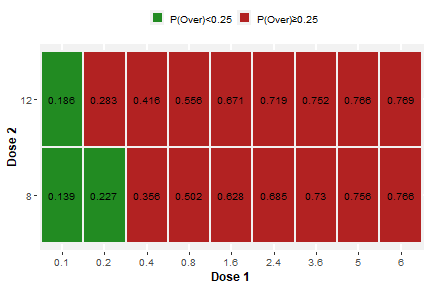
Plot generated by scenario_jointBLRM()
for trial "B"
Note that we supplied an additional argument,
ewoc.threshold, to the call to
scenario_jointBLRM. This argument states the feasibility
bound of the EWOC criterion (cf. Babb et al. (1998)) and is displayed by
the plots to indicate which doses are fulfilling the EWOC criterion (and
could be used for the next cohort). This argument is optional and
defaults to 0.25 if not specified, so that it is actually
not needed unless a different value shall be used as feasibility bound
of the EWOC criterion.
There are further options for customizing plots, e.g. to indicate which type of plots is used for combination therapy trials (heatmap or bar diagram). Detail on this and further arguments for the output plots are specified in the documentation.
One can also enable automatic saving of the posterior summaries by
supplying the optional arguments file.name and
path. If the latter specifies a valid (writable) directory,
the output will be saved as .xlsx file under the specified
file.name. Additionally, one can save the output plots by
further specifying plots.save = TRUE (provided a valid
path and file.name were supplied). In this
case, it is recommended to specify the format of the output plots via
plot.unit, plot.width, and
plot.height. Refer to the documentation for detail on
this.
Evaluating multiple scenarios in parallel
When setting up a prior, one will typically want to evaluate a number
of scenarios at once, but, as Bayesian computation tends to take
relatively long, this may be rather slow for e.g. 10 or more scenarios.
Additionally, one would need e.g. 10 (or more) calls to
scenario_jointBLRM, which may not be practical if the same
scenarios are repeatedly evaluated for slightly altered prior
specifications.
To provide slightly more convenience for these types of exercise, the
function scenario_list_jointBLRM allows to perform multiple
calls to scenario_jointBLRM in parallel for a list of data
scenarios, which both speeds up the computation and simplifies the
evaluation of a number of hypothetical settings. Please note that a
parallel backend needs to be registered to leverage the parallelization,
otherwise the function will be executed sequentially. An example is
provided below.
To use this, consider first a (numbered) list of data scenarios. This
is specified as a list of lists, where each sublist must be of the
format expected by the data argument of
scenario_jointBLRM. For instance, a list with 8 scenarios
could look like this:
data.list = list(
#Scenario 1
list(
dose1 = c( 0.1),
dose2 = c( 0),
n.pat = c( 3),
n.dlt = c( 0),
trial = c("A")),
#Scenario 2
list(
dose1 = c( 0.1),
dose2 = c( 0),
n.pat = c( 3),
n.dlt = c( 1),
trial = c("A")),
#Scenario 3
list(
dose1 = c( 0.1),
dose2 = c( 0),
n.pat = c( 3),
n.dlt = c( 2),
trial = c("A")),
#Scenario 4
list(
dose1 = c( 0.1, 0.2),
dose2 = c( 0, 0),
n.pat = c( 3, 3),
n.dlt = c( 0, 1),
trial = c("A", "A")),
#Scenario 5
list(
dose1 = c( 0.1, 0.2),
dose2 = c( 0, 0),
n.pat = c( 3, 3),
n.dlt = c( 0, 2),
trial = c("A", "A")),
#Scenario 6
list(
dose1 = c( 0.1, 0.2, 0.4),
dose2 = c( 0, 0, 0),
n.pat = c( 3, 3, 3),
n.dlt = c( 0, 0, 1),
trial = c("A", "A", "A")),
#Scenario 7
list(
dose1 = c( 0.1, 0.2, 0.4),
dose2 = c( 0, 0, 0),
n.pat = c( 3, 3, 6),
n.dlt = c( 0, 0, 1),
trial = c("A", "A", "A")),
#Scenario 8
list(
dose1 = c( 0.1, 0.2, 0.4, 0.2),
dose2 = c( 0, 0, 0, 8),
n.pat = c( 3, 3, 6, 3),
n.dlt = c( 0, 0, 1, 0),
trial = c("A", "A", "A", "B"))
)Note that Scenario 8 also contains cohorts from the combination
therapy trial, encoded with the trial name "B" as specified
previously. The cohorts can be given in arbitrary order.
To run the evaluations in parallel, one can additionally register a
paralell backend. There are various existing packages than can achieve
this. A basic construct to register a parallel backend using the
doFuture package is the following
library(doFuture)
#specify number of cores -- here, only 1 for a sequential exacution
n.cores <- 1
#register backend for parallel execution and plan multisession
doFuture::registerDoFuture()
future::plan(future::multisession, workers=n.cores)In this example, only 1 core is selected, which is identical to a
sequential execution. In practice, n.cores can be set
depending on the available processor on which the function is
running.
Now, coming back to the evaluation of scenarios, after registration of the parallel backend one can simply use
results <- scenario_list_jointBLRM(
data.list = data.list,
historical.data = historical.data,
doses.of.interest = doses.of.interest,
dose.ref1 = dose.ref1,
dose.ref2 = dose.ref2,
trials.of.interest = trials.of.interest,
types.of.interest = types.of.interest,
prior.mu = prior.mu,
prior.tau = prior.tau
)
print(results)
#> [[1]]
#> [[1]]$`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.02629 0.06901 0.00000 0.00108 0.24157 0.95389 0.03335 0.01276
#> 0.2+0 0.03834 0.09010 0.00000 0.00245 0.32889 0.92537 0.04987 0.02476
#> 0.4+0 0.05776 0.12113 0.00000 0.00562 0.45832 0.88396 0.06689 0.04915
#> 0.8+0 0.08879 0.16322 0.00000 0.01303 0.62015 0.82253 0.08784 0.08963
#> 1.6+0 0.13789 0.21527 0.00000 0.03152 0.79474 0.73684 0.10943 0.15373
#> 2.4+0 0.17913 0.24919 0.00004 0.05380 0.87765 0.67021 0.12268 0.20711
#> 3.6+0 0.23398 0.28456 0.00024 0.09469 0.93795 0.58855 0.13350 0.27795
#> 5+0 0.29191 0.31325 0.00068 0.15226 0.96966 0.50889 0.13965 0.35146
#> 6+0 0.33007 0.32909 0.00101 0.19635 0.98156 0.46268 0.14039 0.39693
#>
#> [[1]]$`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.09596 0.09113 0.00620 0.07103 0.34521 0.84208 0.12950 0.02842
#> 0.2+8 0.10744 0.10718 0.00681 0.07642 0.41522 0.80799 0.14739 0.04462
#> 0.4+8 0.12558 0.13146 0.00777 0.08460 0.52825 0.76176 0.16513 0.07311
#> 0.8+8 0.15440 0.16589 0.00922 0.09715 0.67305 0.69641 0.18429 0.11930
#> 1.6+8 0.20040 0.21082 0.01139 0.11942 0.82493 0.60701 0.20309 0.18990
#> 2.4+8 0.23946 0.24139 0.01301 0.14203 0.89541 0.53991 0.21173 0.24836
#> 3.6+8 0.29194 0.27425 0.01475 0.18147 0.94769 0.46073 0.21295 0.32632
#> 5+8 0.34776 0.30211 0.01600 0.23634 0.97595 0.38990 0.20507 0.40503
#> 6+8 0.38430 0.31780 0.01583 0.28020 0.98621 0.35231 0.19340 0.45429
#> 0.1+12 0.13536 0.09759 0.02351 0.11183 0.39383 0.70603 0.24972 0.04425
#> 0.2+12 0.14637 0.11139 0.02459 0.11774 0.45308 0.67355 0.26217 0.06428
#> 0.4+12 0.16381 0.13303 0.02590 0.12662 0.55511 0.62862 0.27497 0.09641
#> 0.8+12 0.19172 0.16481 0.02752 0.14010 0.69082 0.56756 0.28587 0.14657
#> 1.6+12 0.23685 0.20790 0.02896 0.16430 0.83609 0.48787 0.28715 0.22498
#> 2.4+12 0.27570 0.23836 0.02865 0.18959 0.90570 0.43039 0.27716 0.29245
#> 3.6+12 0.32825 0.27247 0.02611 0.23329 0.95521 0.36841 0.25258 0.37901
#> 5+12 0.38362 0.30255 0.02177 0.29273 0.98051 0.32100 0.21730 0.46170
#> 6+12 0.41912 0.31983 0.01832 0.33906 0.98942 0.29912 0.19269 0.50819
#>
#>
#> [[2]]
#> [[2]]$`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.26814 0.20897 0.00921 0.21871 0.75773 0.39043 0.27542 0.33415
#> 0.2+0 0.33265 0.22806 0.01631 0.29563 0.81783 0.28592 0.26153 0.45255
#> 0.4+0 0.40530 0.24731 0.02576 0.38520 0.87830 0.20253 0.22777 0.56970
#> 0.8+0 0.47981 0.26293 0.03805 0.48000 0.93033 0.14415 0.18695 0.66890
#> 1.6+0 0.55030 0.27216 0.05140 0.57280 0.96564 0.10492 0.14716 0.74792
#> 2.4+0 0.58816 0.27448 0.06003 0.62448 0.97871 0.08794 0.12830 0.78376
#> 3.6+0 0.62306 0.27480 0.06955 0.67288 0.98712 0.07452 0.11243 0.81305
#> 5+0 0.64911 0.27379 0.07744 0.70944 0.99158 0.06503 0.10179 0.83318
#> 6+0 0.66271 0.27281 0.08181 0.72818 0.99340 0.06048 0.09647 0.84305
#>
#> [[2]]$`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.31803 0.21200 0.03883 0.27016 0.80618 0.27978 0.31595 0.40427
#> 0.2+8 0.37464 0.22757 0.04989 0.33596 0.85582 0.20259 0.28851 0.50890
#> 0.4+8 0.43947 0.24221 0.06374 0.41743 0.90237 0.14130 0.24416 0.61454
#> 0.8+8 0.50804 0.25388 0.07906 0.50597 0.94239 0.09733 0.19520 0.70747
#> 1.6+8 0.57453 0.26152 0.09346 0.59612 0.97093 0.07000 0.15119 0.77881
#> 2.4+8 0.61022 0.26465 0.09964 0.64633 0.98202 0.06004 0.13095 0.80901
#> 3.6+8 0.64223 0.26795 0.10260 0.69411 0.98938 0.05503 0.11411 0.83086
#> 5+8 0.66457 0.27211 0.09871 0.73061 0.99358 0.05467 0.10394 0.84139
#> 6+8 0.67519 0.27561 0.09326 0.74971 0.99529 0.05674 0.09900 0.84426
#> 0.1+12 0.34760 0.20579 0.06406 0.30474 0.81568 0.19935 0.34391 0.45674
#> 0.2+12 0.40176 0.22017 0.07626 0.36739 0.86299 0.14068 0.29892 0.56040
#> 0.4+12 0.46378 0.23381 0.09166 0.44455 0.90747 0.09630 0.24390 0.65980
#> 0.8+12 0.52930 0.24507 0.10814 0.52884 0.94589 0.06590 0.18934 0.74476
#> 1.6+12 0.59243 0.25375 0.11766 0.61408 0.97316 0.04924 0.14498 0.80578
#> 2.4+12 0.62561 0.25906 0.11782 0.66298 0.98376 0.04668 0.12468 0.82864
#> 3.6+12 0.65406 0.26685 0.10897 0.70973 0.99105 0.04985 0.11017 0.83998
#> 5+12 0.67204 0.27716 0.09146 0.74623 0.99504 0.05885 0.10170 0.83945
#> 6+12 0.67941 0.28518 0.07847 0.76665 0.99662 0.06699 0.09851 0.83450
#>
#>
#> [[3]]
#> [[3]]$`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.55392 0.23487 0.10672 0.56626 0.94450 0.05423 0.14813 0.79764
#> 0.2+0 0.62100 0.22417 0.15080 0.65006 0.95843 0.02823 0.09791 0.87386
#> 0.4+0 0.68267 0.21360 0.19681 0.72473 0.97135 0.01569 0.06323 0.92108
#> 0.8+0 0.73574 0.20305 0.24131 0.78820 0.98240 0.00966 0.04240 0.94794
#> 1.6+0 0.77960 0.19223 0.28604 0.83776 0.99039 0.00650 0.02952 0.96398
#> 2.4+0 0.80132 0.18580 0.30996 0.86165 0.99352 0.00502 0.02416 0.97082
#> 3.6+0 0.82049 0.17938 0.33431 0.88245 0.99573 0.00419 0.02007 0.97574
#> 5+0 0.83435 0.17421 0.35317 0.89696 0.99698 0.00354 0.01748 0.97898
#> 6+0 0.84146 0.17137 0.36365 0.90436 0.99751 0.00331 0.01605 0.98064
#>
#> [[3]]$`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.57010 0.23870 0.11961 0.58521 0.95411 0.04638 0.14664 0.80698
#> 0.2+8 0.63093 0.22772 0.16315 0.66154 0.96590 0.02378 0.10065 0.87557
#> 0.4+8 0.68930 0.21501 0.20934 0.73289 0.97661 0.01234 0.06433 0.92333
#> 0.8+8 0.74132 0.20229 0.25455 0.79378 0.98518 0.00676 0.04188 0.95136
#> 1.6+8 0.78449 0.19122 0.29550 0.84357 0.99171 0.00438 0.02938 0.96624
#> 2.4+8 0.80505 0.18657 0.31334 0.86802 0.99443 0.00392 0.02481 0.97127
#> 3.6+8 0.82157 0.18479 0.32088 0.88878 0.99649 0.00436 0.02253 0.97311
#> 5+8 0.83121 0.18717 0.31122 0.90422 0.99773 0.00536 0.02350 0.97114
#> 6+8 0.83470 0.19080 0.29669 0.91165 0.99826 0.00665 0.02486 0.96849
#> 0.1+12 0.58885 0.22947 0.15101 0.60400 0.95623 0.02908 0.13299 0.83793
#> 0.2+12 0.64701 0.21878 0.19374 0.67685 0.96746 0.01465 0.08733 0.89802
#> 0.4+12 0.70278 0.20659 0.23872 0.74507 0.97759 0.00718 0.05475 0.93807
#> 0.8+12 0.75226 0.19485 0.28069 0.80306 0.98590 0.00414 0.03481 0.96105
#> 1.6+12 0.79252 0.18624 0.31502 0.85126 0.99229 0.00310 0.02563 0.97127
#> 2.4+12 0.81058 0.18486 0.32065 0.87471 0.99499 0.00347 0.02347 0.97306
#> 3.6+12 0.82312 0.18922 0.30377 0.89506 0.99708 0.00524 0.02505 0.96971
#> 5+12 0.82759 0.19985 0.26604 0.90954 0.99824 0.00897 0.02914 0.96189
#> 6+12 0.82713 0.20955 0.23260 0.91701 0.99876 0.01278 0.03287 0.95435
#>
#>
#> [[4]]
#> [[4]]$`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.11867 0.11368 0.00285 0.08317 0.42156 0.72987 0.20847 0.06166
#> 0.2+0 0.16832 0.14058 0.00634 0.13091 0.52177 0.57767 0.28551 0.13682
#> 0.4+0 0.23900 0.17944 0.01193 0.19939 0.66692 0.41518 0.30962 0.27520
#> 0.8+0 0.32700 0.22415 0.01969 0.28785 0.82070 0.28735 0.27547 0.43718
#> 1.6+0 0.42030 0.26144 0.02924 0.39036 0.92652 0.19896 0.22597 0.57507
#> 2.4+0 0.47251 0.27665 0.03573 0.45497 0.96065 0.16300 0.19858 0.63842
#> 3.6+0 0.52117 0.28686 0.04252 0.52053 0.97972 0.13457 0.17341 0.69202
#> 5+0 0.55754 0.29191 0.04842 0.57293 0.98840 0.11601 0.15508 0.72891
#> 6+0 0.57648 0.29364 0.05186 0.60143 0.99151 0.10739 0.14621 0.74640
#>
#> [[4]]$`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.18628 0.13471 0.02386 0.15218 0.53638 0.52684 0.33541 0.13775
#> 0.2+8 0.23069 0.15834 0.03157 0.19210 0.63125 0.40788 0.36582 0.22630
#> 0.4+8 0.29222 0.18850 0.04234 0.24976 0.74582 0.28848 0.35725 0.35427
#> 0.8+8 0.36998 0.22202 0.05526 0.32964 0.85779 0.19335 0.30722 0.49943
#> 1.6+8 0.45580 0.25243 0.06800 0.42798 0.93786 0.13186 0.24138 0.62676
#> 2.4+8 0.50490 0.26668 0.07340 0.49072 0.96565 0.11014 0.20534 0.68452
#> 3.6+8 0.55053 0.27857 0.07551 0.55580 0.98266 0.09668 0.17413 0.72919
#> 5+8 0.58374 0.28762 0.07251 0.60975 0.99057 0.09245 0.15219 0.75536
#> 6+8 0.60032 0.29299 0.06827 0.63837 0.99357 0.09346 0.14078 0.76576
#> 0.1+12 0.22155 0.13495 0.04615 0.19109 0.56234 0.38986 0.42474 0.18540
#> 0.2+12 0.26404 0.15625 0.05524 0.22990 0.65119 0.29205 0.42655 0.28140
#> 0.4+12 0.32295 0.18402 0.06725 0.28584 0.75831 0.19990 0.38786 0.41224
#> 0.8+12 0.39743 0.21564 0.08076 0.36222 0.86533 0.13197 0.31501 0.55302
#> 1.6+12 0.47953 0.24583 0.09131 0.45613 0.94205 0.09378 0.23702 0.66920
#> 2.4+12 0.52616 0.26162 0.09122 0.51759 0.96925 0.08418 0.19670 0.71912
#> 3.6+12 0.56861 0.27725 0.08313 0.58244 0.98505 0.08485 0.16344 0.75171
#> 5+12 0.59812 0.29156 0.06918 0.63659 0.99265 0.09321 0.14154 0.76525
#> 6+12 0.61195 0.30077 0.05775 0.66487 0.99528 0.10149 0.13108 0.76743
#>
#>
#> [[5]]
#> [[5]]$`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.24977 0.15939 0.02780 0.22110 0.62143 0.34596 0.37606 0.27798
#> 0.2+0 0.33372 0.17764 0.05234 0.31479 0.71227 0.18186 0.34897 0.46917
#> 0.4+0 0.43305 0.20155 0.08178 0.42542 0.82251 0.09285 0.24353 0.66362
#> 0.8+0 0.53212 0.22284 0.11275 0.53840 0.91761 0.05181 0.16035 0.78784
#> 1.6+0 0.61830 0.23384 0.14357 0.64406 0.96940 0.03166 0.10776 0.86058
#> 2.4+0 0.66103 0.23540 0.16184 0.69932 0.98398 0.02440 0.08762 0.88798
#> 3.6+0 0.69831 0.23419 0.17950 0.74859 0.99171 0.01909 0.07211 0.90880
#> 5+0 0.72486 0.23168 0.19371 0.78465 0.99524 0.01579 0.06265 0.92156
#> 6+0 0.73833 0.22983 0.20204 0.80272 0.99649 0.01458 0.05739 0.92803
#>
#> [[5]]$`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.30531 0.17914 0.05170 0.27246 0.72103 0.23654 0.37864 0.38482
#> 0.2+8 0.37687 0.19477 0.07589 0.35207 0.79578 0.13470 0.32380 0.54150
#> 0.4+8 0.46261 0.20991 0.10754 0.45071 0.87398 0.06770 0.23520 0.69710
#> 0.8+8 0.55273 0.22218 0.14098 0.55828 0.93669 0.03485 0.15359 0.81156
#> 1.6+8 0.63459 0.22940 0.17217 0.66147 0.97464 0.02033 0.10184 0.87783
#> 2.4+8 0.67517 0.23175 0.18515 0.71663 0.98638 0.01719 0.08428 0.89853
#> 3.6+8 0.70923 0.23440 0.19008 0.76608 0.99316 0.01686 0.07215 0.91099
#> 5+8 0.73142 0.23858 0.18203 0.80303 0.99632 0.01876 0.06757 0.91367
#> 6+8 0.74133 0.24245 0.17331 0.82163 0.99746 0.02162 0.06618 0.91220
#> 0.1+12 0.33556 0.17496 0.07780 0.30647 0.73527 0.15761 0.39127 0.45112
#> 0.2+12 0.40400 0.18910 0.10365 0.38218 0.80682 0.08480 0.31265 0.60255
#> 0.4+12 0.48600 0.20300 0.13593 0.47605 0.88037 0.04084 0.21272 0.74644
#> 0.8+12 0.57204 0.21483 0.16929 0.57851 0.94061 0.02066 0.13542 0.84392
#> 1.6+12 0.64954 0.22356 0.19358 0.67816 0.97648 0.01398 0.09136 0.89466
#> 2.4+12 0.68696 0.22877 0.19789 0.73125 0.98777 0.01420 0.07767 0.90813
#> 3.6+12 0.71663 0.23699 0.18356 0.78005 0.99423 0.01835 0.07137 0.91028
#> 5+12 0.73360 0.24843 0.15551 0.81524 0.99711 0.02646 0.07069 0.90285
#> 6+12 0.73972 0.25748 0.13210 0.83393 0.99815 0.03410 0.07095 0.89495
#>
#>
#> [[6]]
#> [[6]]$`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.05911 0.06818 0.00063 0.03493 0.24966 0.91423 0.07783 0.00794
#> 0.2+0 0.09033 0.08776 0.00215 0.06284 0.32539 0.82261 0.15384 0.02355
#> 0.4+0 0.14489 0.12307 0.00565 0.11133 0.45913 0.64384 0.26451 0.09165
#> 0.8+0 0.23176 0.18129 0.01172 0.18675 0.67629 0.43971 0.30121 0.25908
#> 1.6+0 0.34174 0.24353 0.01977 0.29132 0.87424 0.28997 0.26181 0.44822
#> 2.4+0 0.40748 0.27144 0.02508 0.36426 0.94082 0.23060 0.22948 0.53992
#> 3.6+0 0.46949 0.29098 0.03067 0.44250 0.97390 0.18543 0.19892 0.61565
#> 5+0 0.51563 0.30114 0.03564 0.50778 0.98694 0.15717 0.17738 0.66545
#> 6+0 0.53948 0.30490 0.03865 0.54403 0.99138 0.14457 0.16604 0.68939
#>
#> [[6]]$`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.13106 0.09683 0.01567 0.10728 0.37996 0.70805 0.24788 0.04407
#> 0.2+8 0.16116 0.11591 0.02119 0.13268 0.46257 0.60029 0.31297 0.08674
#> 0.4+8 0.21043 0.14603 0.02984 0.17527 0.58846 0.45174 0.36967 0.17859
#> 0.8+8 0.28621 0.18952 0.04152 0.24147 0.75561 0.30389 0.35784 0.33827
#> 1.6+8 0.38493 0.23782 0.05464 0.33850 0.90060 0.19503 0.29274 0.51223
#> 2.4+8 0.44619 0.26221 0.06073 0.40829 0.95072 0.15516 0.24764 0.59720
#> 3.6+8 0.50464 0.28184 0.06339 0.48516 0.97848 0.13016 0.20631 0.66353
#> 5+8 0.54764 0.29489 0.06066 0.55205 0.98990 0.12065 0.17545 0.70390
#> 6+8 0.56929 0.30157 0.05736 0.58970 0.99353 0.11880 0.15963 0.72157
#> 0.1+12 0.16861 0.10178 0.03547 0.14699 0.42186 0.55455 0.37150 0.07395
#> 0.2+12 0.19744 0.11826 0.04245 0.17231 0.49480 0.45280 0.42021 0.12699
#> 0.4+12 0.24468 0.14532 0.05306 0.21371 0.61089 0.32741 0.43966 0.23293
#> 0.8+12 0.31742 0.18591 0.06541 0.27818 0.76967 0.21384 0.38958 0.39658
#> 1.6+12 0.41223 0.23284 0.07658 0.37137 0.90767 0.14039 0.29564 0.56397
#> 2.4+12 0.47085 0.25807 0.07755 0.44007 0.95526 0.11903 0.24141 0.63956
#> 3.6+12 0.52609 0.28065 0.07099 0.51750 0.98145 0.11237 0.19308 0.69455
#> 5+12 0.56553 0.29818 0.05881 0.58437 0.99198 0.11701 0.16055 0.72244
#> 6+12 0.58456 0.30825 0.04911 0.62246 0.99526 0.12328 0.14559 0.73113
#>
#>
#> [[7]]
#> [[7]]$`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.04107 0.04924 0.00043 0.02345 0.17909 0.96468 0.03372 0.00160
#> 0.2+0 0.06287 0.06327 0.00156 0.04278 0.23383 0.91703 0.07808 0.00489
#> 0.4+0 0.10298 0.09030 0.00423 0.07765 0.33752 0.78440 0.18784 0.02776
#> 0.8+0 0.17426 0.14502 0.00872 0.13480 0.54760 0.57015 0.28477 0.14508
#> 1.6+0 0.27667 0.21783 0.01473 0.21921 0.80490 0.38519 0.28245 0.33236
#> 2.4+0 0.34253 0.25489 0.01879 0.28040 0.90423 0.30748 0.25768 0.43484
#> 3.6+0 0.40680 0.28307 0.02328 0.34997 0.95731 0.24851 0.22889 0.52260
#> 5+0 0.45561 0.29927 0.02711 0.41235 0.97881 0.21277 0.20469 0.58254
#> 6+0 0.48110 0.30597 0.02927 0.44877 0.98594 0.19524 0.19310 0.61166
#>
#> [[7]]$`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.11369 0.08215 0.01366 0.09462 0.32360 0.77678 0.20059 0.02263
#> 0.2+8 0.13562 0.09562 0.01829 0.11356 0.37909 0.69106 0.26402 0.04492
#> 0.4+8 0.17336 0.12013 0.02564 0.14466 0.48312 0.55616 0.34117 0.10267
#> 0.8+8 0.23644 0.16176 0.03527 0.19611 0.65544 0.39261 0.37683 0.23056
#> 1.6+8 0.32773 0.21739 0.04580 0.27414 0.84625 0.25927 0.33264 0.40809
#> 2.4+8 0.38855 0.24900 0.05067 0.33365 0.92292 0.20827 0.28655 0.50518
#> 3.6+8 0.44901 0.27590 0.05227 0.40395 0.96624 0.17497 0.23832 0.58671
#> 5+8 0.49494 0.29417 0.05008 0.46720 0.98430 0.15997 0.20190 0.63813
#> 6+8 0.51859 0.30336 0.04690 0.50519 0.99022 0.15582 0.18457 0.65961
#> 0.1+12 0.15221 0.08991 0.03285 0.13417 0.37594 0.61827 0.33582 0.04591
#> 0.2+12 0.17321 0.10106 0.03870 0.15281 0.42447 0.53061 0.39281 0.07658
#> 0.4+12 0.20941 0.12245 0.04733 0.18385 0.51609 0.41087 0.44202 0.14711
#> 0.8+12 0.27006 0.16073 0.05769 0.23394 0.67608 0.28076 0.42922 0.29002
#> 1.6+12 0.35803 0.21441 0.06621 0.31123 0.85821 0.18857 0.34417 0.46726
#> 2.4+12 0.41660 0.24654 0.06669 0.37020 0.93059 0.15941 0.28329 0.55730
#> 3.6+12 0.47438 0.27603 0.05989 0.44150 0.97139 0.14850 0.22411 0.62739
#> 5+12 0.51736 0.29837 0.04842 0.50651 0.98789 0.15118 0.18277 0.66605
#> 6+12 0.53878 0.31060 0.04001 0.54503 0.99295 0.15636 0.16319 0.68045
#>
#>
#> [[8]]
#> [[8]]$`trial-A`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+0 0.03306 0.04007 0.00032 0.01892 0.14553 0.98176 0.01781 0.00043
#> 0.2+0 0.05178 0.05267 0.00120 0.03502 0.19503 0.95110 0.04730 0.00160
#> 0.4+0 0.08759 0.07862 0.00340 0.06522 0.29536 0.84184 0.14363 0.01453
#> 0.8+0 0.15450 0.13497 0.00728 0.11586 0.51412 0.62893 0.26020 0.11087
#> 1.6+0 0.25497 0.21270 0.01266 0.19266 0.79321 0.43361 0.27358 0.29281
#> 2.4+0 0.32107 0.25277 0.01640 0.25071 0.90094 0.34750 0.25593 0.39657
#> 3.6+0 0.38635 0.28352 0.02024 0.31869 0.95758 0.28067 0.23195 0.48738
#> 5+0 0.43636 0.30143 0.02354 0.38032 0.97980 0.24004 0.20898 0.55098
#> 6+0 0.46261 0.30893 0.02567 0.41675 0.98673 0.22006 0.19818 0.58176
#>
#> [[8]]$`trial-B`
#> mean sd q.2.5% q.50% q.97.5% P([0,0.16)) P([0.16,0.33)) P([0.33,1])
#> 0.1+8 0.09138 0.06342 0.01104 0.07747 0.25118 0.86816 0.12625 0.00559
#> 0.2+8 0.10884 0.07303 0.01509 0.09309 0.29139 0.79879 0.18753 0.01368
#> 0.4+8 0.14135 0.09474 0.02144 0.12001 0.38402 0.66708 0.28553 0.04739
#> 0.8+8 0.20064 0.14033 0.03054 0.16548 0.57193 0.48151 0.36203 0.15646
#> 1.6+8 0.29220 0.20583 0.03981 0.23645 0.81067 0.32176 0.33846 0.33978
#> 2.4+8 0.35528 0.24340 0.04414 0.29253 0.90788 0.25585 0.29750 0.44665
#> 3.6+8 0.41885 0.27509 0.04554 0.35960 0.96243 0.21052 0.25397 0.53551
#> 5+8 0.46762 0.29589 0.04289 0.42272 0.98343 0.18912 0.21588 0.59500
#> 6+8 0.49297 0.30599 0.04019 0.46165 0.98977 0.18203 0.19545 0.62252
#> 0.1+12 0.12938 0.07330 0.02905 0.11540 0.30764 0.72073 0.26244 0.01683
#> 0.2+12 0.14614 0.08079 0.03421 0.13107 0.34354 0.63959 0.32973 0.03068
#> 0.4+12 0.17738 0.09910 0.04212 0.15809 0.42293 0.50796 0.41295 0.07909
#> 0.8+12 0.23449 0.14065 0.05171 0.20333 0.59697 0.35318 0.43979 0.20703
#> 1.6+12 0.32294 0.20356 0.05913 0.27372 0.82331 0.23534 0.36822 0.39644
#> 2.4+12 0.38392 0.24123 0.05927 0.32939 0.91729 0.19522 0.30568 0.49910
#> 3.6+12 0.44512 0.27499 0.05315 0.39748 0.96792 0.17594 0.24301 0.58105
#> 5+12 0.49138 0.29939 0.04260 0.46209 0.98670 0.17329 0.19647 0.63024
#> 6+12 0.51482 0.31231 0.03513 0.50208 0.99235 0.17616 0.17270 0.65114
#>
#>
#> attr(,"rng")
#> attr(,"rng")[[1]]
#> [1] 10407 2133139108 -773071531 -244150766 -1207652757 1191582384 1240354545
#>
#> attr(,"doRNG_version")
#> [1] "1.7.4"Here, except for the first argument, data.list, we have
provided precisely the same arguments as for the call to
scenario_jointBLRM from the previous section. This would
also work for all other (optional) arguments of
scenario_jointBLRM, except for file.name.
Instead of this argument, scenario_list_jointBLRM uses the
optional argument file.names (notice the plural), which has
the same function as file.name for
scenario_jointBLRM, but needs to contain a vector with a
file name for each scenario instead. As before, the argument is ignored
unless you want to save the output and have supplied a
path.
Note further that the historical data provided to
scenario_list_jointBLRM is included in the posterior
evaluations for every scenario, i.e., the historical data is interpreted
in the same fashion as for scenario_jointBLRM, and does not
vary across the scenarios in the list.
To obtain a prior configuration, it is now recommended to create a sufficiently large number of scenarios, and repeatedly evaluate the scenarios to gradually alter the prior distribution and obtain an appropriate setup and behavior in the considered scenarios. However, as mentioned before, one will probably not be able to consider every potential scenario in advance, so that this is mostly a heuristic check and is not guaranteed to yield an optimal prior. Due to this, further evaluation of the prior should be performed to confirm that it shows appropriate operating characteristics in the long-run. This will be the subject of the following section.
Simulating dose-finding trials with sim_jointBLRM
Simulations of trials usually aim to estimate the operating characteristics (OC) of a dose-finding trial design, which includes e.g. the ratio of MTDs declared on each dose (or on doses with DLT rate in a specific dosing interval), the number of patients per dose and the number of DLTs in the trial, as well as further metrics for the simulation results.
To estimate the operating characteristics of the BLRM, one usually uses simulated trials. That is, one assumes a fixed set of available dose levels and a fixed dose-toxicity scenario – i.e. a specification of the DLT rate for each available dose level – and simulates a number of trials in this setting. This allows to e.g. estimate the average number of MTDs in the correct interval or the number of patients treated at overly toxic doses for this specific dose-toxicity scenario.
As it is of course not possible to test all possible dose-toxicity scenarios, one will typically confine oneself to a selected number of scenarios that illustrate different possible cases. These should usually range from scenarios that follow the prior expectations, over more extreme scenarios that assume the involved drugs to be considerably more (or less) toxic than expected by the prior, up to scenarios that deliberately assume the trials to result in heterogeneous data or that the true dose-toxicity relationship does not follow the logistic model of the BLRM (i.e. one assumes the BLRM to be misspecified).
As an example how such simulations can be performed via
sim_jointBLRM, we will again consider the setting of the
previous section. For this, recall that a monotherapy and combination
therapy trial shall run (partially) in parallel, where we wanted to
consider the dose levels given in the variable
doses.of.interest and use the data from a historical trial
as given in the historical.data argument.
The function sim_jointBLRM supports simulation of
(parallel) monotherapy or combination therapy trials. Internally, it is
differentiated between three types, namely monotherapy for compound 1
("mono1"), monotherapy for compound 2
("mono2"), and combination therapy with compounds 1 and 2
("combi"). For each of these types, the function can
actively simulate 2 trials (i.e., six parallel trials in total), and the
order in which cohorts are enrolled in each of the up to six trials can
be specified freely. The simulated trials can be activated independently
of each other, i.e. one can choose how many of the parallel trials are
simulated.
Additionally, historical data from arbitrarily many trials that are already concluded can be given, and furthermore, data that was observed in the actively simulated trials before the start of the simulation can be included as well (which allows to simulate how one or more trials might continue after already having observed a specific data scenario).
Naming conventions for sim_jointBLRM
The function features a large number of optional parameters to allow relatively flexible customization of the simulated trials, e.g. regarding the number of patients per cohort, criteria for MTD selection, escalation rules, and further topics. Refer to the documentation for more detail and the parameters that are not discussed in this vignette.
To allow some settings to deviate across the simulated trials, many
of the arguments of sim_jointBLRM exist in six variations,
one for each simulated trial. To differentiate between these, the
arguments feature trial-specific suffixes as given in the following
table.
| Trial | Suffix of function arguments |
|---|---|
| 1st monotherapy trial for compound 1 | mono1.a |
| 1st monotherapy trial for compound 2 | mono2.a |
| 1st combination therapy trial | combi.a |
| 2nd monotherapy trial for compound 1 | mono1.b |
| 2nd monotherapy trial for compound 2 | mono2.b |
| 2nd combination therapy trial | combi.b |
Here, the first and second trial for a specific trial type mean
simply that these are independently escalating trials of the same type,
but does not imply a specific order or some other dependency across the
first and second trial (except that they are jointly modeled in the
BLRM). It is for instance possible to just simulate the
mono2.b trial and deactivate all other trials.
The take away message from these naming conventions is that one needs to use the parameter with the corresponding suffix when an specific option shall only affect one specific trial but not the others. Arguments that do not have a suffix will in general always affect all simulated trials.
Apart from the argument names, the suffixes for the six potentially simulated trials are also used to refer to the corresponding trial when the order in which the trials enroll cohorts is specified. Furthermore, they are also used to encode the trials when historical data that was previously recorded in an actively simulated trial shall be included. Refer to the documentation for more detail.
Configuring the trial simulations
First and foremost, one needs to decide which of the possible six
trials need to be simulated and activate the corresponding trials. For
the example in this vignette, an monotherapy trial for compound 1 and a
combination therapy trial shall be included. We will assume that we have
chosen to utilize the mono1.a and combi.a
trials to represent these trials (although it would be equivalent to use
the mono1.b and combi.b trials or even a
combination of mono1.b and combi.a). To
activate the trials, we can supply the arguments
active.mono1.a = TRUE
active.combi.a = TRUENow, we specify the corresponding available dose levels. For this, we will use the same doses as specified previously:
doses.mono1.a = c(0.1, 0.2, 0.4, 0.8, 1.6, 2.4, 3.6, 5, 6)
doses.combi.a = rbind(
rep(doses.mono1.a, times=2),
rep(c(8, 12), each = length(doses.mono1.a)))Note that doses.mono1.a is a vector, while
doses.combi.a needs to be a matrix with two rows, so that
each column specifies a dose combination to be considered in the
combination therapy trial. The code above generated every possible
combination of the doses from the monotherapy trial for compound 1 with
the doses 8 and 12 for compound 2.
As for the function scenario_jointBLRM, one again needs
to specify reference doses and a prior distribution for the BLRM. For
this, the same argument names and formats as for
scenario_jointBLRM are used, i.e. the arguments
dose.ref1, dose.ref2, prior.mu,
and prior.tau can be used in the form that was specified in
the previous section. The same holds for the historical data, for which
we will assume that the same observations as before are used, namely
historical.data
#> $dose1
#> [1] 0 0 0 0 0
#>
#> $dose2
#> [1] 2 4 8 12 16
#>
#> $n.pat
#> [1] 3 3 3 9 12
#>
#> $n.dlt
#> [1] 0 0 0 1 2
#>
#> $trial
#> [1] "H1" "H1" "H1" "H1" "H1"Additionally, we need to provide starting dose levels for each activated trials. Note that these must be elements of the set of available dose levels, otherwise the function will report an error. We will assume that the monotherapy trial will start with the lowest available dose, 0.1, which is specified via
start.dose.mono1.a = 0.1We will later assume additionally that the monotherapy trial enrolls at least 3 or 4 cohorts before the first combination therapy cohort is processed, and due to this, we assume the combination trial to use a slightly larger starting dose. For instance, suppose that the combination therapy shall begin with the dose combination of 0.2 of compound 1 and 8 of compound 2. This is expressed by using
start.dose.combi.a1 = 0.2
start.dose.combi.a2 = 8Note that two arguments need to be specified for this, with suffix
.combi.a1 for the first compound and .combi.a2
for the second compound. Both of these arguments belong to the
combi.a trial, while the endings .a1 and
.a2 are used to differentiate between the setting for the
first and second compound of the combi.a trial. They are
also used for other arguments that may need different settings for the
two combination therapy partners in the same combination therapy
trial.
Optional general specification
As a next step, the general course of the trial can be defined, consisting of the number of patients per cohort, the maximum number of patients in the trial, and the order in which cohorts are enrolled. All of these arguments are optional, and relatively sensible defaults will be used when they are not specified.
First, to set the number of patients per cohort, one can use e.g.
cohort.size = 3Note that cohort.size is the global variant of this
argument, i.e. it affects all simulated trials. If it is not provided
explicitly, the function will use the default value of 3. If a specific
trial shall deviate from this value, one can use the trial-specific
parameter with the corresponding trial suffix, e.g.
cohort.size.mono1.b = 4Note that this would only affect the mono1.b trial,
which we do not plan to simulate in the current example. For the
mono1.a and combi.a trial, which we do want to
simulate, the global cohort size of 3 is still in effect. The same would
apply if cohort.size is not given to the function, as the
default value would have been used in this case.
One can also provide a vector of multiple cohort sizes, in which case
the function will randomly select one of the given cohort sizes for each
cohort. The cohort sizes will then be drawn uniformly at random from the
possible numbers specified in ‘cohort.size’ if no further options are
given. If it is desired to draw each cohort size with a specific
probability, one can use the optional argument cohort.prob
to provide the probability for each cohort size, e.g. by setting
In this example, the mono1.b trial would select the size
of each cohort randomly according to the corresponding probabilities,
i.e. a cohort size of 2 is drawn with a probability of 0.05, a size of 3
with probability 0.85, and a cohort size of 4 will be chosen with a
probability of 0.1. As before, there is a global and trial-specific
version of the argument cohort.prob.
As a next step, we want to set the maximum number of patients for each trial. We will assume the maximum number of patients to be 36 for the combination therapy trial and 45 for the monotherapy trial. This can be set via the trial-specific arguments
max.n.mono1.a = 45
max.n.combi.a = 36Note that there is also a global argument max.n, which
will not be used in this example.
Now, the order in which cohorts are enrolled is specified. For this,
the function uses the argument cohort.queue, which
provides, as the name says, a queue of cohorts that is processed during
the simulation. This can be done by supplying a simple pattern which is
automatically repeated (note however that the function will drop a
warning in this case) or by supplying a complex pattern that already
contains every cohort that may potentially be arriving throughout the
trial. For instance, to simply simulate cohorts for mono1.a
and combi.a alternately, one can use
cohort.queue = c("mono1.a", "combi.a")This order would then be repeated until both trials concluded due to being stopped early, having found an MTD, or reaching the maximum patient number.
The cohort queue is mostly used to enforce a specific order of the cohorts in a trial. For this, consider again the example from the previous section, where a monotherapy and combination trial shall be simulated in parallel. Assume for instance that one wants to start the combination therapy trial after the first four monotherapy cohorts were processed, and, afterwards, the trials shall enroll cohorts alternately. This is expressed by setting the queue to
cohort.queue = c("mono1.a", "mono1.a", "mono1.a", rep(c("mono1.a", "combi.a"),
times=12))
head(cohort.queue, 9)
#> [1] "mono1.a" "mono1.a" "mono1.a" "mono1.a" "combi.a" "mono1.a" "combi.a" "mono1.a" "combi.a"Note that c("mono1.a", "combi.a") is repeated 12 times,
which translates to 12 "combi.a" and 15
"mono1.a" cohorts in the queue. As the cohort size was
previously set to 3 patients, this results in a total of 36 patients for
combi.a in the queue and 45 patients for
mono1.a, i.e. precisely the maximum sample size that was
specified. This ensures that the function will not need to repeat the
queue automatically. Note that it would not be a problem to provide a
cohort queue containing more patients than required, as the function
will simply ignore cohorts for each simulated trial once it has
concluded. Therefore, it is also possible to use e.g.
Here, we supplied much more cohorts than needed (100 for combination therapy), which is still equivalent to the previous queue. Only supplying lower numbers of cohorts will cause the function to operate differently, as it will repeat the cohort queue until all trials concluded.
Note that the trials in the cohort queue can also be indicated with numbers (which usually leads to shorter code than the full trial names), as stated in the following table.
| Trial | Suffix of function arguments | Number |
|---|---|---|
| 1st monotherapy trial for compound 1 | mono1.a |
1 |
| 1st monotherapy trial for compound 2 | mono2.a |
2 |
| 1st combination therapy trial | combi.a |
3 |
| 2nd monotherapy trial for compound 1 | mono1.b |
4 |
| 2nd monotherapy trial for compound 2 | mono2.b |
5 |
| 2nd combination therapy trial | combi.b |
6 |
Hence, the previous cohort queue could be equivalently written as
Escalation decisions in simulted trials
As a next step, we specify rules for dose escalation decisions. As mentioned before, the function will by default apply EWOC-based escalation, which means that only doses can be chosen for which the posterior probability of overdosing is below 0.25. This boundary could also be altered, by using the argument
ewoc.threshold = 0.25As EWOC only states an upper boundary, the function will afterwards
maximize the probability of having a DLT rate within the target interval
among the doses that satisfy EWOC. If this still leads to multiple
possible doses, the function will subsequently maximize the dose of the
first compound among the doses with the same probability of target
dosing. To maximize the second compound instead, one can set the
argument esc.comp.max, which may have the values
1 or 2 and indicates which component the
function will maximize in case of draws. Note that one can also deviate
from maximizing the target probability and maximize directly the dose
among the levels that satisfy EWOC, but this is not covered in this
vignette. Refer to the documentation of the argument
esc.rule for information on this.
Neuenschwander et al. (2014) recommend
to impose a maximum increment of the dose additionally to EWOC, in order
to avoid overly large dosing steps during dose escalations. This can be
realized by using the esc.step family of arguments, which
is available as a global variant and six trial-specific parameters. The
escalation step is interpreted as the maximum factor of dose escalation.
For instance, when an escalation step is set to the value 2, the dose
can at most be doubled in each escalation decision. If no escalation
step is given, the function will set it to the maximum factor between
subsequent dose levels as an additional constrain on the escalation.
However, there might be situations in which one may not want a fixed maximum factor between escalations, e.g. if a factor cannot express the increments between planned dose levels. Consider for instance a trial with planned doses of 10, 20, 30, and 40 mg. In order to allow escalating from 10 to 20, the escalation step would need to be set to 2, but this would also allow escalating from 20 to 40 without enrolling patients at 30. To prevent this, one can set
esc.constrain = TRUEThis argument constrains escalations to the given set of dose levels,
i.e. only the next larger prespecified dose can be used. If activated,
esc.constrain causes the function to ignore any given
escalation step and always restrict escalation decisions to the next
larger dose (but does not pose a boundary when deescalating the dose).
As before, esc.constrain is the global variant, while
additional arguments like esc.constrain.mono1.b only affect
the corresponding trial.
MTD selection
It now remains to specify the rules for MTD selection in the trial.
There are two different arguments that control how the function
selects the MTD of a simulated trial, namely mtd.decision
and mtd.enforce. Both are available as global arguments
without suffix and as trial-specific argument with the corresponding
suffix. The argument mtd.decision is a named list with five
entries of the form
mtd.decision = list(
min.dlt = 1,
pat.at.mtd = 6,
min.pat = 12,
target.prob = 0.5,
rule = 2
)The list stated above is also the default argument. The first four
list entries correspond to the demanded boundaries of four different
conditions that are used to determine an MTD. The last entry,
mtd.decision$rule does not pose a condition itself, but
dictates which of the other conditions are mandatory instead.
During the simulation, the MTD can only be determined as the dose
that was used for the last cohort, which is always the current candidate
for the MTD. For MTD selection, the function will then first demand that
the current dose level is also recommended for the next cohort again
(so-called stabilization criterion). If this is the case, it will check
whether the current dose also satisfies the conditions determined by
mtd.decision, and, if these are satisfied, declare the
current dose to be the MTD of this trial. The precise conditions and the
corresponding entries in mtd.decision are listed in the
following table.
| No. | List entry | Condition |
|---|---|---|
| 1 | min.dlt |
At least min.dlt patients with DLTs were
observed in the trial. |
| 2 | pat.at.mtd |
At least pat.at.mtd patients were treated
with the current dose. |
| 3 | min.pat |
At least min.pat patients were treated in
the current trial. |
| 4 | target.prob |
The probability that the current dose has DLT rate in
the target interval is at least target.prob. |
In terms of the effect of the entry rule, the following
to possibilities are supported
If
ruleis1: Conditions 1 and 2 are necessary, and at least one of conditions 3 and 4 must be satisfied as well.If
ruleis2: Conditions 1 and 3 are necessary, and at least one of conditions 2 and 4 must be satisfied as well.
Other decision rule specifications may be supported in the future.
The second argument that affects MTD selection,
mtd.enforce, is a logical value that controls how the
function behaves when a trial reaches the specified maximum sample size.
By default, mtd.enforce is FALSE, which causes
trials that reach the maximum sample size to be considered as not having
found an MTD, which will also be noted in the output. If the parameter
is instead TRUE, the current dose will be considered the
MTD when the maximum sample size is reached before the conditions for
MTD selections are satisfied. This can e.g. be used if one wants to
approximate the average dosing recommendations after a specific number
of cohorts based on simulations. For instance, when
max.n = 15, mtd.enforce = TRUE, one can set
mtd.decision to values that cannot be reached within 15
patients (e.g., mtd.decision$rule = 2 and
mtd.decision$min.pat = 16), so that the function will
simply report the dosing recommendation after 15 patients as the MTD and
not apply the MTD decision rule. This option can also be set for
individual trials (via the parameters with the corresponding
suffix).
Dose-toxicity scenarios and trial simulations
It remains to specify a dose-toxicity scenario before the simulation can be started. That is, a DLT rate for each dose level is assumed, which is then used by the function to simulate the number of patients experiencing DLT in each cohort.
The arguments for specifying the DLT rate in one of the simulated
trials are named according to the pattern tox.[...], where
[...] can be any of the trial-specific suffixes. Note that
there is no global variant of this argument. For the situation discussed
in the previous chapters, the required arguments are therefore
tox.mono1.a and tox.combi.a.
First, recall the specification of the dose levels for the monotherapy trial:
doses.mono1.a
#> [1] 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5.0 6.0The DLT rates (or toxicities) must therefore be specified for 9 dose
levels. This is done by supplying a vector of this length, so that the
entry tox.mono1.a[i] specifies the DLT rate of the dose
given in doses.mono1.a[i]. In our case, this could
e.g. be
#Compound 1: 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6
tox.mono1.a = c(0.01, 0.02, 0.03, 0.05, 0.09, 0.12, 0.19, 0.29, 0.43)Similarly, for the combination therapy trial, the dose levels were given as
doses.combi.a
#> [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12] [,13] [,14] [,15] [,16] [,17] [,18]
#> [1,] 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6
#> [2,] 8.0 8.0 8.0 8.0 8.0 8.0 8.0 8 8 12.0 12.0 12.0 12.0 12.0 12.0 12.0 12 12Here, each column specifies a dose, so that the argument
tox.combi.a needs one entry for each column, i.e. 18
entries in our example. Similarly to before, entry i of the
argument provides the DLT rate of the dose given in column
i of doses.combi.a. Hence, a dose-toxicity
scenario for this case could be
#Compound 1: 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6
#compound 2: 8 8 8 8 8 8 8 8 8 12 12 12 12 12 12 12 12 12
tox.combi.a = c(0.02, 0.03, 0.04, 0.08, 0.14, 0.21, 0.32, 0.39, 0.47, 0.04, 0.06, 0.09, 0.12, 0.17, 0.29, 0.45, 0.52, 0.61)Note that one will usually consider multiple different dose toxicity scenarios to assess the performance of the prior in different settings and situations. In the following, we will only consider the scenario specified above for simplicity.
This completes the setup and specification of the trials that are to
be simulated. As a last step, it remains to specify the number of
simulated studies via the parameter n.studies. As an
example, 1 study will be used in the following. Note however that this
is of course by far not sufficient for a proper evaluation of the prior,
where one should simulate something around the order of e.g. 1000 to
10000 studies. However, be aware that depending on the specifications,
each study will require up to multiple minutes of run time for the
repeated calls to Stan, so that 1000 or more trials may take multiple
hours or even nearly a day to simulate. It is therefore recommended to
run the simulation in parallel.
If a cluster is available, a parallel backend consisting of multiple
nodes with multiple cores can be registered, so that the function can
leverage the capabilities of a cluster with a nested foreach loop. This
is however purely optional. Further, when running simulations in
parallel with a moderate number of cores, it is recommended to consider
the argument working.path in the documentation, as this
allows to save a reload MCMC results across nodes and cores.
At last, one can also optionally specify the arguments
path and file.name, which will cause the
function to automatically write the summarized simulation results to a
.xlsx file of the specified name under the specified path.
Otherwise, the simulation results are only returned to R.
Putting all of the previous explanations together, the call to
sim_jointBLRM would therefore be:
sim_result <- sim_jointBLRM(
active.mono1.a = TRUE,
active.combi.a = TRUE,
doses.mono1.a = doses.mono1.a,
doses.combi.a = doses.combi.a,
dose.ref1 = dose.ref1,
dose.ref2 = dose.ref2,
start.dose.mono1.a = start.dose.mono1.a,
start.dose.combi.a1 = start.dose.combi.a1,
start.dose.combi.a2 = start.dose.combi.a2,
tox.mono1.a = tox.mono1.a,
tox.combi.a = tox.combi.a,
historical.data = historical.data,
prior.mu = prior.mu,
prior.tau = prior.tau,
max.n.mono1.a = max.n.mono1.a,
max.n.combi.a = max.n.combi.a,
cohort.size = cohort.size,
cohort.queue = cohort.queue,
ewoc.threshold = ewoc.threshold,
esc.constrain = esc.constrain,
mtd.decision = mtd.decision,
n.studies = 1
)
print(sim_result)
#> $`results combi.a`
#> underdose target dose overdose max n reached before MTD all doses too toxic
#> Number of trials 0 1 0 0 0
#> Percentage 0 100 0 0 0
#> Percentage not all too toxic 0 100 0 0 NA
#>
#> $`summary combi.a`
#> Median Mean Min. Max. 2.5% 97.5%
#> #Pat underdose 9.0000000 9.0000000 9.0000000 9.0000000 9.0000000 9.0000000
#> #Pat target dose 6.0000000 6.0000000 6.0000000 6.0000000 6.0000000 6.0000000
#> #Pat overdose 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
#> #Pat (all) 15.0000000 15.0000000 15.0000000 15.0000000 15.0000000 15.0000000
#> #DLT underdose 1.0000000 1.0000000 1.0000000 1.0000000 1.0000000 1.0000000
#> #DLT target dose 1.0000000 1.0000000 1.0000000 1.0000000 1.0000000 1.0000000
#> #DLT overdose 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
#> #DLT (all) 2.0000000 2.0000000 2.0000000 2.0000000 2.0000000 2.0000000
#> % overdose 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
#> % DLT 0.1333333 0.1333333 0.1333333 0.1333333 0.1333333 0.1333333
#>
#> $`MTDs combi.a`
#> 0.1+8 0.2+8 0.4+8 0.8+8 1.6+8 2.4+8 3.6+8 5+8
#> Dose "0.1+8" "0.2+8" "0.4+8" "0.8+8" "1.6+8" "2.4+8" "3.6+8" "5+8"
#> True P(DLT) "0.02" "0.03" "0.04" "0.08" "0.14" "0.21" "0.32" "0.39"
#> True category "underdose" "underdose" "underdose" "underdose" "underdose" "target dose" "target dose" "overdose"
#> MTD declared (n) "0" "0" "0" "0" "0" "0" "0" "0"
#> 6+8 0.1+12 0.2+12 0.4+12 0.8+12 1.6+12 2.4+12 3.6+12
#> Dose "6+8" "0.1+12" "0.2+12" "0.4+12" "0.8+12" "1.6+12" "2.4+12" "3.6+12"
#> True P(DLT) "0.47" "0.04" "0.06" "0.09" "0.12" "0.17" "0.29" "0.45"
#> True category "overdose" "underdose" "underdose" "underdose" "underdose" "target dose" "target dose" "overdose"
#> MTD declared (n) "0" "0" "0" "0" "0" "0" "1" "0"
#> 5+12 6+12
#> Dose "5+12" "6+12"
#> True P(DLT) "0.52" "0.61"
#> True category "overdose" "overdose"
#> MTD declared (n) "0" "0"
#>
#> $`#pat. combi.a`
#> 0.1+8 0.2+8 0.4+8 0.8+8 1.6+8 2.4+8 3.6+8 5+8 6+8 0.1+12 0.2+12 0.4+12 0.8+12
#> Dose "0.1+8" "0.2+8" "0.4+8" "0.8+8" "1.6+8" "2.4+8" "3.6+8" "5+8" "6+8" "0.1+12" "0.2+12" "0.4+12" "0.8+12"
#> mean #pat "0" "3" "0" "0" "0" "0" "0" "0" "0" "0" "0" "3" "3"
#> median #pat "0" "3" "0" "0" "0" "0" "0" "0" "0" "0" "0" "3" "3"
#> min #pat "0" "3" "0" "0" "0" "0" "0" "0" "0" "0" "0" "3" "3"
#> max #pat "0" "3" "0" "0" "0" "0" "0" "0" "0" "0" "0" "3" "3"
#> 1.6+12 2.4+12 3.6+12 5+12 6+12
#> Dose "1.6+12" "2.4+12" "3.6+12" "5+12" "6+12"
#> mean #pat "3" "3" "0" "0" "0"
#> median #pat "3" "3" "0" "0" "0"
#> min #pat "3" "3" "0" "0" "0"
#> max #pat "3" "3" "0" "0" "0"
#>
#> $`#DLT combi.a`
#> 0.1+8 0.2+8 0.4+8 0.8+8 1.6+8 2.4+8 3.6+8 5+8 6+8 0.1+12 0.2+12 0.4+12 0.8+12
#> Dose "0.1+8" "0.2+8" "0.4+8" "0.8+8" "1.6+8" "2.4+8" "3.6+8" "5+8" "6+8" "0.1+12" "0.2+12" "0.4+12" "0.8+12"
#> mean #DLT "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "1"
#> median #DLT "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "1"
#> min #DLT "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "1"
#> max #DLT "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "1"
#> 1.6+12 2.4+12 3.6+12 5+12 6+12
#> Dose "1.6+12" "2.4+12" "3.6+12" "5+12" "6+12"
#> mean #DLT "0" "1" "0" "0" "0"
#> median #DLT "0" "1" "0" "0" "0"
#> min #DLT "0" "1" "0" "0" "0"
#> max #DLT "0" "1" "0" "0" "0"
#>
#> $`results mono1.a`
#> underdose target dose overdose max n reached before MTD all doses too toxic
#> Number of trials 0 1 0 0 0
#> Percentage 0 100 0 0 0
#> Percentage not all too toxic 0 100 0 0 NA
#>
#> $`summary mono1.a`
#> Median Mean Min. Max. 2.5% 97.5%
#> #Pat underdose 21.0000000 21.0000000 21.0000000 21.0000000 21.0000000 21.0000000
#> #Pat target dose 6.0000000 6.0000000 6.0000000 6.0000000 6.0000000 6.0000000
#> #Pat overdose 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
#> #Pat (all) 27.0000000 27.0000000 27.0000000 27.0000000 27.0000000 27.0000000
#> #DLT underdose 1.0000000 1.0000000 1.0000000 1.0000000 1.0000000 1.0000000
#> #DLT target dose 2.0000000 2.0000000 2.0000000 2.0000000 2.0000000 2.0000000
#> #DLT overdose 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
#> #DLT (all) 3.0000000 3.0000000 3.0000000 3.0000000 3.0000000 3.0000000
#> % overdose 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
#> % DLT 0.1111111 0.1111111 0.1111111 0.1111111 0.1111111 0.1111111
#>
#> $`MTDs mono1.a`
#> 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5
#> Dose "0.1" "0.2" "0.4" "0.8" "1.6" "2.4" "3.6" "5"
#> True P(DLT) "0.01" "0.02" "0.03" "0.05" "0.09" "0.12" "0.19" "0.29"
#> True category "underdose" "underdose" "underdose" "underdose" "underdose" "underdose" "target dose" "target dose"
#> MTD declared (n) "0" "0" "0" "0" "0" "0" "1" "0"
#> 6
#> Dose "6"
#> True P(DLT) "0.43"
#> True category "overdose"
#> MTD declared (n) "0"
#>
#> $`#pat. mono1.a`
#> 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6
#> Dose "0.1" "0.2" "0.4" "0.8" "1.6" "2.4" "3.6" "5" "6"
#> mean #pat "3" "3" "3" "6" "3" "3" "6" "0" "0"
#> median #pat "3" "3" "3" "6" "3" "3" "6" "0" "0"
#> min #pat "3" "3" "3" "6" "3" "3" "6" "0" "0"
#> max #pat "3" "3" "3" "6" "3" "3" "6" "0" "0"
#>
#> $`#DLT mono1.a`
#> 0.1 0.2 0.4 0.8 1.6 2.4 3.6 5 6
#> Dose "0.1" "0.2" "0.4" "0.8" "1.6" "2.4" "3.6" "5" "6"
#> mean #DLT "0" "0" "0" "1" "0" "0" "2" "0" "0"
#> median #DLT "0" "0" "0" "1" "0" "0" "2" "0" "0"
#> min #DLT "0" "0" "0" "1" "0" "0" "2" "0" "0"
#> max #DLT "0" "0" "0" "1" "0" "0" "2" "0" "0"
#>
#> $`historical data`
#> Obs1 Obs2 Obs3 Obs4 Obs5
#> Dose 1 0 0 0 0 0
#> Dose 2 2 4 8 12 16
#> N Pat. 3 3 3 9 12
#> N DLT 0 0 0 1 2
#> Trial 7 7 7 7 7
#>
#> $prior
#> mean SD
#> mu_a1 -0.7081851 3.0000000
#> mu_b1 0.0000000 1.0000000
#> mu_a2 -0.7081851 3.0000000
#> mu_b2 0.0000000 1.0000000
#> mu_eta 0.0000000 1.1210000
#> tau_a1 -1.3862944 0.3536465
#> tau_b1 -2.0794415 0.3536465
#> tau_a2 -1.3862944 0.3536465
#> tau_b2 -2.0794415 0.3536465
#> tau_eta -2.0794415 0.3536465
#>
#> $specifications
#> Value -
#> seed "558535265" "-"
#> dosing.intervals "0.16" "0.33"
#> esc.rule "ewoc" "-"
#> esc.comp.max "1" "-"
#> dose.ref1 "6" "-"
#> dose.ref2 "12" "-"
#> saturating "FALSE" "-"
#> ewoc.threshold "0.25" "-"
#> start.dose.mono1.a "0.1" "-"
#> esc.step.mono1.a "2" "-"
#> esc.constrain.mono1.a "TRUE" "-"
#> max.n.mono1.a "45" "-"
#> cohort.size.mono1.a "3" "-"
#> cohort.prob.mono1.a "1" "-"
#> mtd.decision.mono1.a$target.prob "0.5" "-"
#> mtd.decision.mono1.a$pat.at.mtd "6" "-"
#> mtd.decision.mono1.a$min.pat "12" "-"
#> mtd.decision.mono1.a$min.dlt "1" "-"
#> mtd.decision.mono1.a$rule "2" "-"
#> mtd.enforce.mono1.a "FALSE" "-"
#> backfill.mono1.a "FALSE" "-"
#> backfill.size.mono1.a "3" "-"
#> backfill.prob.mono1.a "1" "-"
#> backfill.start.mono1.a "0.1" "-"
#> start.dose.combi.a1 "0.2" "-"
#> start.dose.combi.a2 "8" "-"
#> esc.step.combi.a1 "2" "-"
#> esc.step.combi.a2 "1.5" "-"
#> esc.constrain.combi.a1 "TRUE" "-"
#> esc.constrain.combi.a2 "TRUE" "-"
#> max.n.combi.a "36" "-"
#> cohort.size.combi.a "3" "-"
#> cohort.prob.combi.a "1" "-"
#> mtd.decision.combi.a$target.prob "0.5" "-"
#> mtd.decision.combi.a$pat.at.mtd "6" "-"
#> mtd.decision.combi.a$min.pat "12" "-"
#> mtd.decision.combi.a$min.dlt "1" "-"
#> mtd.decision.combi.a$rule "2" "-"
#> mtd.enforce.combi.a "FALSE" "-"
#> backfill.combi.a "FALSE" "-"
#> backfill.size.combi.a "3" "-"
#> backfill.prob.combi.a "1" "-"
#> backfill.start.combi.a1 "0.1" "-"
#> backfill.start.combi.a2 "8" "-"
#>
#> $`Stan options`
#> Value
#> chains 4.0
#> iter 13500.0
#> warmup 1000.0
#> adapt_delta 0.8
#> max_treedepth 15.0This completes the setup of the simulation function for prior evaluations. As mentioned before, it is recommended to evaluate multiple different scenarios for at least multiple hundred studies when assessing the performance of an actual design.
References
Session info
sessionInfo()
#> R version 4.1.2 (2021-11-01)
#> Platform: x86_64-w64-mingw32/x64 (64-bit)
#> Running under: Windows 10 x64 (build 19044)
#>
#> Matrix products: default
#>
#> locale:
#> [1] LC_COLLATE=German_Germany.1252 LC_CTYPE=German_Germany.1252 LC_MONETARY=German_Germany.1252
#> [4] LC_NUMERIC=C LC_TIME=German_Germany.1252
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] future.batchtools_0.12.0 parallelly_1.35.0 doRNG_1.8.2 rngtools_1.5.2
#> [5] doFuture_1.0.0 future_1.32.0 foreach_1.5.2 decider_0.0.0.9011
#>
#> loaded via a namespace (and not attached):
#> [1] httr_1.4.2 RcppParallel_5.1.5 StanHeaders_2.21.0-7 askpass_1.1 highr_0.9
#> [6] stats4_4.1.2 base64url_1.4 yaml_2.2.2 progress_1.2.2 globals_0.16.1
#> [11] pillar_1.7.0 backports_1.4.1 glue_1.6.1 digest_0.6.29 checkmate_2.1.0
#> [16] colorspace_2.0-3 htmltools_0.5.2 pkgconfig_2.0.3 rstan_2.21.5 listenv_0.8.0
#> [21] purrr_0.3.4 scales_1.2.1 processx_3.8.1 brew_1.0-6 openxlsx_4.2.5.2
#> [26] tibble_3.1.6 openssl_1.4.6 generics_0.1.1 farver_2.1.0 ggplot2_3.3.6
#> [31] ellipsis_0.3.2 cachem_1.0.6 withr_2.5.0 cli_3.4.1 magrittr_2.0.2
#> [36] crayon_1.5.1 memoise_2.0.1 evaluate_0.14 ps_1.6.0 fs_1.5.2
#> [41] fansi_1.0.2 doParallel_1.0.17 pkgbuild_1.3.1 tools_4.1.2 loo_2.5.1
#> [46] data.table_1.14.2 prettyunits_1.1.1 hms_1.1.1 lifecycle_1.0.3 matrixStats_0.62.0
#> [51] stringr_1.4.0 munsell_0.5.0 zip_2.2.0 callr_3.7.3 pkgdown_2.0.7
#> [56] compiler_4.1.2 rlang_1.0.6 grid_4.1.2 iterators_1.0.14 rstudioapi_0.13
#> [61] rappdirs_0.3.3 labeling_0.4.2 rmarkdown_2.11 gtable_0.3.0 codetools_0.2-18
#> [66] flock_0.7 inline_0.3.19 R6_2.5.1 gridExtra_2.3 knitr_1.37
#> [71] dplyr_1.0.7 fastmap_1.1.0 future.apply_1.9.1 utf8_1.2.2 rprojroot_2.0.3
#> [76] desc_1.4.1 stringi_1.7.6 parallel_4.1.2 Rcpp_1.0.8.3 vctrs_0.4.2
#> [81] batchtools_0.9.17 tidyselect_1.1.1 xfun_0.29